Polüskleroos ehk hulgiskleroos ehk hulgikoldekõvastumus ehk multipleksskleroos (ladina keeles sclerosis multiplex; inglise keeles multiple sclerosis; lühend MS või SM) on peamiselt inimestel esinev krooniline, kogu elu kestev, peamiselt tuvastamata põhjustega, harvaesinev haigus, millele on iseloomulikud muutused kesknärvisüsteemi valgeaines.
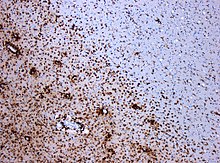
Demüelisatsioon polüskleroosi korral. Värvunud CD68 kude näitab haiguskolde piirkonnas mitmeid
makrofaage. Mõõtkava 1:100
Eestis põeb 2011. aasta seisuga sclerosis multiplex'i ligi 1500 inimest.
EtioloogiaAutoimmuunhaigusena
Arvatakse, et polüskleroosi kui autoimmuunhaiguse käigus hävitab autoimmuunne põletik närvirakkude ümber oleva müeliinkihi. Vahel võib esineda ka närvirakkude jätkete katkemine.
Autoimmuunsusega seostatakse lümfoid(-immuun)süsteemi homöostaasi häirumist ja nende rakkude tegevuse tulemusel tekkivaid koldeid pea- ja seljaajus.
Polüskleroos mõjutab pea- ja seljaaju närvirakkude võimet omavahel suhelda. Kommunikatsioon toimub elektriliste signaalide ehk aktsioonipotentsiaalide levimisel piki aksoneid, mis on kaetud erilise isoleeriva müeliinikihiga.
Polüskleroosi korral ei suuda organismi vastavad rakud eristada omi rakke võõrastest ja tekivad immuunreaktsioonid, mille tulemusel ründab organismi immuunsüsteem oma närvisüsteemi ja kahjustub müeliin. Polüskleroosi põdevate haigete organismis stimuleerivad leukotsüüdid (T-lümfotsüüdid, B-lümfotsüüdid ja makrofaagid jt) rünnakut närvirakke isoleeriva müeliinkesta vastu .
Patogeneesis mängivad olulist rolli mikrogliia ja T-rakud.
Müeliinkihi kahjustusel võib tulemuseks olla närviimpulsside liikumise aeglustumine või lakkamine, mis omakorda on närvisüsteemile laastava tagajärjega . Kahjustused tekivad mitmete väikeste kolletena pea- ja seljaaju valgeaines (valgeaine koosneb peamiselt müeliinist). Väliselt jätavad kolded mulje, nagu oleks tegemist tihenenud piirkondadega. Sellest tuleneb haiguse nimetus sclerosis multiplex – mitmesed tihenenud kolded .
Hormoonid ja hormoonidelaadset toimet avaldavad ained
Polüskleroos ja D-vitamiini vaegus
Uuritakse D-vitamiini ja/või selle vaeguse ning seotud geenide rolli haiguse tekkimisel ja progresseerumisel, kuna uuringutes on täheldatud, et seerumi D-vitamiini tase võib teatud haigetel näidata haiguse ägenemisega seonduvat.
Päriliku haigusena
Haiguse täpne etioloogia ei ole teada, aga arvatakse, et oluline on teatud pärilike ja keskkonnategurite koosmõju.
Polüskleroos ei ole pärilik haigus, kuid võrreldes tavapopulatsiooniga on polüskleroosiga inimeste lastel mõnevõrra suurem risk haigestuda. Uurimuste andmetel on polüskleroosihaige inimese perekonnaliikmete haigestumise risk järgmine:
- õel 4,4%
- vennal 3,2%
- lapsel 1,8%
- kui SM-i põevad mõlemad vanemad, suureneb risk järglasel ligikaudu 20%.
Assotsiatsiooniuuringutes on leitud ka kandidaatgeene, mida seostatakse sclerosis multiplexiga. Kuuendas kromosoomis paikneva leukotsüütide antigeense süsteemi (HLA geenikompleksi alleele DR15 ja DQ6) seostatakse suurenenud haigestumise riskiga (HLA-C554 ja HLA-DRB1 lookusel on kaitsev efekt). Uurijad ei saa siiski kogu haiguse teket seletada HLA süsteemi geneetiliste muutustega. Hiljuti avastati kahes Euroopa riigis (Saksamaa ja Prantsusmaa) SM-i populatsioonis kaks uut üksiku nukleotiidi polümorfismi (SNP) IL2RA geenis (rs12722489, rs2104286) ja üks SNP IL7RA geenis (rs6897932), mida uurijad seostavad haiguse tekkega .
Suitsetamine võib geneetilise soodumusega inimestel suurendada riski haigestuda polüskleroosi.
Viirused
Geneetilise soodumusega inimestel võivad SM-i põhjustada ka mitmed viirused. Kuigi nende täpne roll pole seni veel selge, ollakse seisukohal, et enamik SM-i patsiente on nakatunud Epsteini-Barri viirusega, lisaks seostatakse haigust veel ka Herpes simplex'i viiruse, punetise-, leetri- ja mumpsiviirusega.
EpidemioloogiaHaigus tabab tavaliselt noori inimesi ja on naistel kaks korda sagedasem kui meestel. Esinemissagedus on 2–150 inimesel 100 000 kohta. Eestis on vastav näitaja umbes 100 inimest 100 000 kohta.
Polüskleroosi on kõige sagedasem noori täiskasvanuid invaliidistav neuroloogiline haigus. Haiguse tekkeiga jääb tavaliselt 10. ja 60. eluaasta vahele. Kõige rohkem haigestub 20–30-aastasi inimesi. Eestis on umbes 1500 sclerosis multiplex'i diagnoosiga patsienti ja terves maailmas umbes 2 miljonit. Polüskleroosi haigestumise risk oleneb ka sellest, millisel laiuskraadil on isik elanud enne 15-aastaseks saamist – parasvöötme laiuskraadidel on haigestumus suurem. Riskirühmaks on naised ja sagedamini haigestuvad valgenahalised inimesed kui asiaadid ja mustanahalised.
SümptomidSõltuvalt kohast, kus kahjustuskolded on tekkinud, võib inimesel esineda mitmesuguseid kaebusi.
Sümptomid:
Füüsilise koormuse või kõrgenenud välistemperatuuri mõjul võivad teatud haigussümptomid süveneda ja seda nimetatakse Uhthoffi fenomeniks. Polüskleroosil on eri raskusastmetega haigusvorme. Umbes 80% juhtudest on tegemist nn tavalise vormiga, kus haigus kulgeb ägenemiste ja vaibumistega. Umbes 20% juhtudest on haigus primaarselt progresseeruva kuluga, mis on pahaloomuline ravile mittealluv vorm. Haigusnähud süvenevad kiiresti ja keskmiselt viie aasta pärast kujuneb invaliidsus. Pooltel patsientidel, kellel esineb ägenemiste ja remissioonidega kulg, areneb sekundaarselt progresseeruv vorm.
Sclerosis multiplex'i alatüübid
Haiguskulu järgi eristatakse nelja sclerosis multiplexi vormi:
- Ägenemiste ehk retsidiivide ja remissioonidega SM (remissioon on stabiilne seisund, kus ägenemise ajal tekkinud sümptomid on vähenenud või kadunud)
See on kõige sagedasem polüskleroosi vorm, mis esineb umbes 40% kõigist haigusjuhtudest. Ägenemiste ja remissioonidega SM-i vormile on iseloomulikud ägenemised, mis tekivad iseenesest ja mööduvad (täielikult või osaliselt) teatud aja jooksul, kuni seisund stabiliseerub. Selle vormi puhul ei esine SM-ile iseloomulike nähtude progresseerumist ehk sümptomid ei süvene ilma uute ägenemisteta.
- Sekundaarse progresseerumisega SM
- ägenemistega
- ägenemisteta
Kui ägenemiste ja remissioonidega SM-i haiged ei saa ravi, areneb 10 aasta jooksul ligikaudu pooltel nendest välja sekundaarselt progresseeruv polüskleroosi vorm. Seda tõenäosust saab märgatavalt vähendada, kui alustada võimalikult kiiresti ravi (preparaatidega, mis modifitseerivad haiguse kulgu, nn immunomodulleerivat ravi) pärast diagnoosi panekut. Osal sekundaarselt progresseeruva SM-iga haigetel võivad tekkida uued ägenemised, osal aga mitte. Sekundaarse progresseerumise korral süvenevad (pikkamööda) haigussümptomid ja töövõimetuse aste.
- Primaarselt progresseeruv SM
Primaarselt progresseeruva polüskleroosi haigetele on haiguse diagnoosi panekust alates omane sümptomite pidev järkjärguline süvenemine ilma uute retsidiivideta. See on haigusvorm, kus ei esine selgete ägenemiste ega paranemistega perioode. See haigusvorm esineb kõige sagedamini inimestel vanuses 40 aastat ja vanemad (harvadel juhtudel haigestuvad sellesse SM-i vormi ka nooremad inimesed). Erinevalt teistest SM-i vormidest esineb progresseeruvat vormi naistel ja meestel võrdselt. Haiguse esmasteks tundemärkideks on kõnnakuhäired, mis raskendavad oluliselt liikumist.
Diagnoos ja raviSclerosis multiplex'i diagnoos, mis tuleks vormistada ladinakeelsena, on kliiniline ja radioloogiline. Eristatakse kliiniliselt kindlat ja laboratoorselt kindlalt, kliiniliselt võimalikku ning laboratoorselt võimalikku diagnoosi, mille aluseks on kindlad diagnoosi kriteeriumid.
SM diagnoosimiseks peab olema tõestatud magnetresonantstomograafilisel ülesvõttel ajust (magnetresonantstomograafia MRT-s) dissemineeritud kahjustus ajas ja ruumis, kliinilised sümptomid peavad diagnoosimiseks kestma vähemalt 24 tundi ning kahe haigusataki (ägenemise) vahele peab jääma vähemalt 1 kuu.
Lisaks kliinilistele sümptomitele on väga olulised ka radioloogilised ja laboratoorsed andmed: pea- ja seljaaju MRT uuring ning oligoklonaalse gammapaatia määramine liikvorist. KT ehk röntgenkompuutertomograafia on peale MRT teine tänapäevane piltdiagnostikameetod. Kuid diagnoosimisel on see kasutatav ainult üksikutel juhtudel ja pigem diferentsiaaldiagnostilistel eesmärkidel teiste haiguste välistamiseks, samuti juhul, kui MRT-d pole võimalik teha (vastunäidustused MRT-ks).
Polüskleroosi ei ole võimalik tänapäeval olemasoleva raviga välja ravida. On olemas ravimite grupid – beetainterferoonid (tsütokiinid) ja kopaksoon, mis vähendavad ägenemiste arvu 1/3 võrra. Vahel kasutatakse ka keemiaravipreparaate, kuid seda otsustatakse alati individuaalselt, sõltuvalt haiguse kulust ja vormist. Üldisteks ravipõhimõteteks on hoiduda liigsest füüsilisest koormusest ja kuumusest. Kuum ilm ja palavik võivad esile kutsuda närvijuhtivuse aeglustumist ning raskendada sümptomeid.
AjaluguSclerosis multiplex on üks paljudest haigustest, mis eksisteeris kaua enne omale nime saamist. Arstid usuvad, et juba keskajast pärinevad kirjutised teatud sümptomitest, mis viitavad SM-le. Üks esimesi teadaolevaid polüskleroosi põdevaid patsiente oli Hollandist pärit naine Lidwina, kes suri 1433. aastal. Pariisi Ülikooli professor, dr Jean-Martin Charcot, keda kutsutakse ka "neuroloogia isaks", oli esimene, kes kirjeldas sclerosis multiplexi kui haigust. Tema ühel naissoost patsiendil esines ebaharilik sümptomite kombinatsioon. Ühestki dr Jean-Martin Charcot' kasutatud ravimeetodist polnud kasu. Pärast patsiendi surma lahkas ta naise aju ning avastas ajus haiguskolded. Ta nimetas need sclerose en plaques'iks. Müeliin avastati peagi pärast seda, kuid tema olulisus haiguse tekkes jäi veel kauaks teadmata. Haiguse kulgu mõjutavad ravimid võeti kliinilises praktikas kasutusele alles 1990. aastate keskel
 CD-d ehk CD-molekulid täidavad organismis erinevaid ülesandeid, osaledes nii signaalrakkude, retseptoritena, osad vajavad diferenseerumiseks ensüümide kaasabi ja osad markerid leiavad kasutust kliinilises meditsiinis.
CD-d ehk CD-molekulid täidavad organismis erinevaid ülesandeid, osaledes nii signaalrakkude, retseptoritena, osad vajavad diferenseerumiseks ensüümide kaasabi ja osad markerid leiavad kasutust kliinilises meditsiinis.











